DockOnSurf execution is controlled by editing an input file. In this file,
all mandatory and optional parameters are specified using a keyword = value pair.
An example of such a file is provided in dockonsurf/examples/dockonsurf.inp.
Although none of the three stages is compulsory for a DockOnSurf execution, the
necessary keywords for each of the stages will be also considered as mandatory.
They are mandatory in the case of running such stage.
For some keywords, the use of more than one value is possible. In order to
separate such values, a whitespace (” “), a comma (“,”) or a semicolon (“;”) can
all be used. Some values need to be specified in groups (eg. each of the three
vectors of the cell matrix). In such case groups are defined by enclosing the
elements around parentheses-like characters: “()”, “[]” or “{}”. (eg. “(a1,
a2,a3) [b1 b2 b3] {c1;c2; c3}”).
Global
Parameters in this section have an effect on the entire DockOnSurf execution.
batch_q_sys (mandatory): The scheduler managing the job submission in
the computing center. If set to “False” DockOnSurf will perform a dry run,
where all structures and the directory hierarchy will be created but no job
will be submitted/run.
Accepted values: “SGE”, “LSF”, “SLURM” or “False”.
code (mandatory): The program to carry out the geometry optimizations.
Accepted values: “CP2K”, “VASP”, “dftb+” or “mace”.
max_jobs (optional): The maximum number of jobs in a certain status:
running (“r”), pending/queued (“p” or “q”), or the sum of both (“rp” or “rq”).
Accepted values: positive integers together to the letters defining the
status (eg. “7r”, “5p” or “10r 5q 12rq”), or “False”. Combination of different
values is possible. Default value: False
pbc_cell (optional): The cell to which Periodic Boundary conditions is
going to be applied. It’s used in the detection of collisions and dissociation
of hydrogen atoms.
Accepted values: The cell matrix: (a1 a2 a3) (b1 b2 b3) (c1 c2 c3) or
“False”. Default value: False
Note
When using VASP as code, the PBC cell must be provided either through
the pbc_cell keyword or inside the coordinates file. (eg. POSCAR/CONTCAR). Similarly when using DFTB+ the PBC cell can be provided either through the pbc_cell keyword (automatically using a supercell format), but it can also be given through the dft_in.hsd input file through a gen format file, or with the explicit coordinates description.
potcar_dir (optional): The path to the directory containing all the
elements with their corresponding POTCAR files.
Accepted values: The path of a directory (eg. /home/cmarti/potcars/) or
“False”. Default value: False
model_mace (optional): The path to mace model to be use.
Accepted values: The path of a file (eg. /home/cmarti/
*.model) or
“False”. Default value: False
project_name (optional): The name of the project. It will appear in the
job names of the submitted calculations.
Accepted values: all. Default value: nothing
run_type (mandatory): Type of run that you want to perform.
It will activate the execution of each of the different stages.
Accepted values: Isolated, Screening, Refinement, Full = Isolated +
Screening + Refinement, Adsorption = Screening + Refinement. More than
one value can be specified separated by : Isolated Screening
special_atoms (optional): Non-standard atom types defined in the
coordinates file together with the standard atom it is related to grouped
inside parentheses.
Accepted values: groups of parentheses containing each a couple of
non-standard, standard chemical symbol. (eg. (Fe1 Fe) (Fe2 Fe) (O1 O)),
or “False”. Default value: False
subm_script (keyword-dependent): The script for the job submission.
Mandatory if batch_q_sys is not set to “False”.
Accepted values: The name/path of the file.
Screening
Parameters in this section have an effect on the Screening stage.
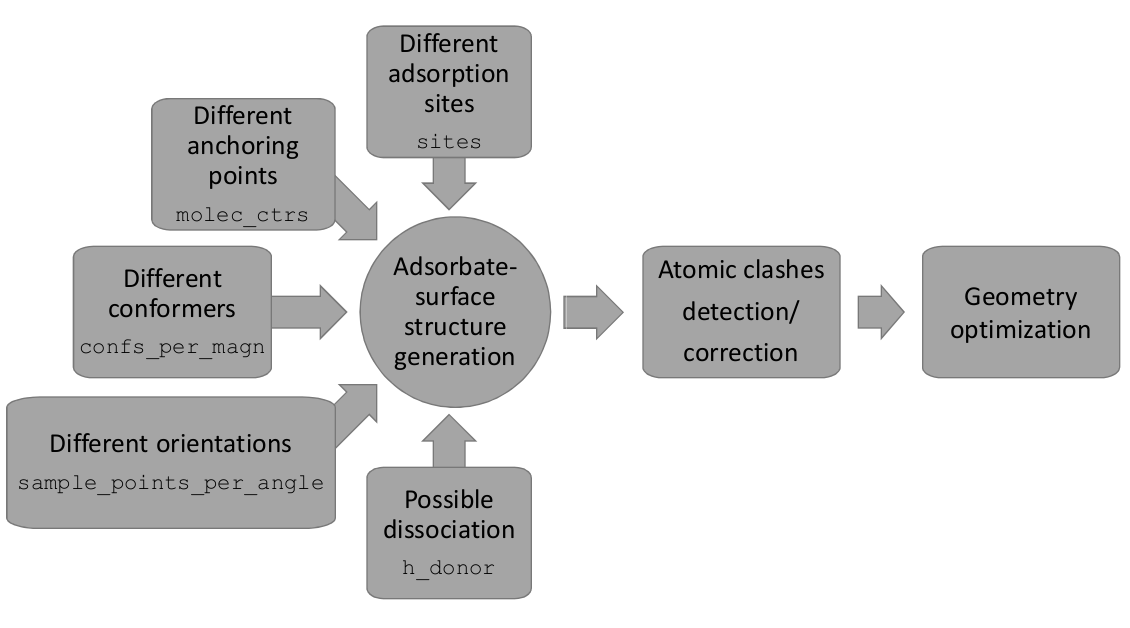
Legend: Schematic overview for the screening (main) step of DockOnSurf with the related keywords in the input file written in verbatim.
adsorption_height (optional): The height at which the adsorbate should
be placed above the surface. Units: Ångstroms.
Accepted values: Positive decimal number. Default Value: 2.5
collision_threshold (optional): When detecting atomic clashes with the
collision of spheres method, collision_threshold sets the coefficient by
which the covalent radius of each atom should be multiplied to get the sphere
radius. When set to False the detection of atomic clashes with the collision
of spheres method is not carried out.
Accepted values: Positive decimal number or “False”. Default Value: False
Warning
If both, the collision_threshold and min_coll_height are set to
“False”, atomic clashes will NOT be checked.
confs_per_magn (optional): The number of conformers to select per
magnitude/quantity. It selects the conformers with most different values of
the relevant quantity. If the moment of inertia (MOI) is chosen as magnitude,
in select_magns,
setting confs_per_magn to 1 picks the conformer with the largest value of
moment of inertia, setting it to 2 picks the ones having the largest and the
smallest, setting it to three, the largest, the median and the smallest, and
so on. If energy is chosen as magnitude, setting confs_per_magn to 1 picks
the most stable conformer (the smallest value of energy), setting it to 2
picks the ones having the largest and the smallest, setting it to three, the
largest, the median and the smallest, and so on.
Accepted values: Positive integers.
exclude_ads_ctr (optional): Whether to exclude the adsorption
center/anchoring point from the atomic clashes detection.
Accepted values: “True” or “False”. Default Value: False
h_acceptor (optional): Which atom types/indices of the surface act as
proton acceptors.
Accepted values: Chemical symbols of surface atoms, atom indices of surface
atoms or “all”. The use of more than one value is possible. Default Value:
all.
h_donor (optional): Which atom types/indices of the adsorbate act as
proton donors. When set to “False” dissociation of protons is disabled.
Accepted values: Chemical symbols of adsorbate atoms, atom indices of
adsorbate atoms or “False”. The use of more than one value is possible.
Default Value: False.
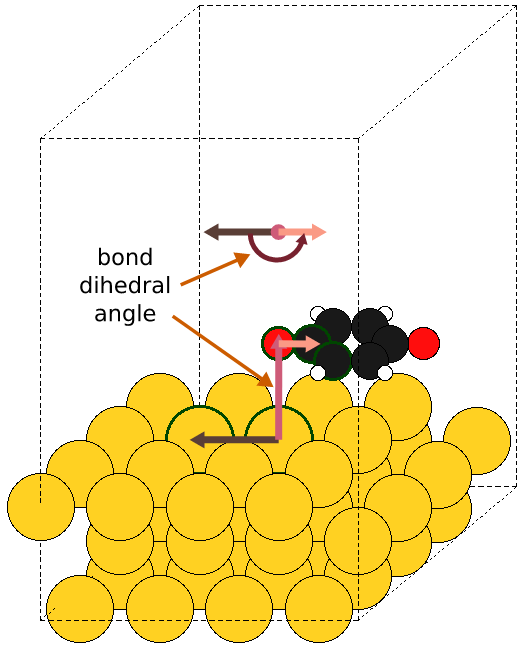
max_helic_angle (optional): The maximum value for the helicopter
rotation of the adsorbate over the surface in degrees (see figure underneath).
Accepted values: Positive decimal number. Default Value: 180.0.
max_structures (optional): Maximum number of adsorbate-surface
structures to generate. Structures are chosen randomly.
Accepted values: Positive integer or “False”.
Warning
When max_structures is set, structures are chosen randomly and
reproducibility is not kept.
min_coll_height (optional): The minimum height of atoms above the
surface for them to be considered as non-colliding with the surface. It can
only be used if the surface normal is one of the cartesian axes (both positive
and negative).
When set to False the detection of atomic clashes with the minimum height
method is not carried out.
Accepted values: Positive decimal number or “False”. Default Value: 1.5
Warning
If both the collision_threshold and min_coll_height are set to
“False”, atomic clashes will NOT be checked.
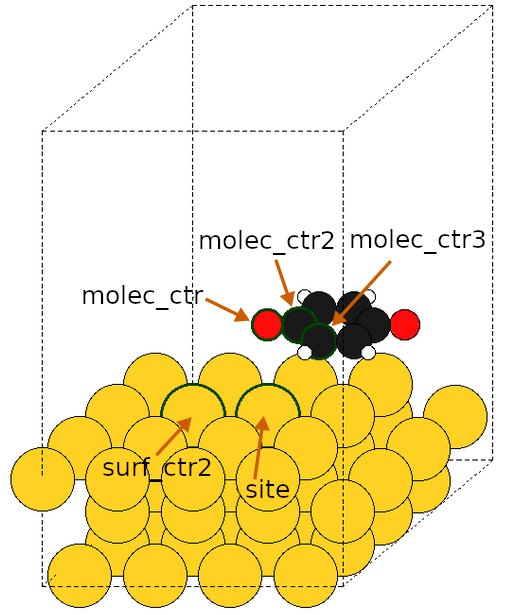
molec_ctrs (mandatory): The (groups of) atom indices in the
adsorbate to be used as adsorption centers/anchoring points (ie. the centers
to be placed right above the surface site). When a group of atom indices is
defined by enclosing them inside parentheses-like characters (“()”, “[]” or
“{}”), the adsorption center/anchoring point is defined as the average of the
atom’s coordinates. It is useful to define π-modes like in ethylene or
benzene.
Accepted values: atom indices of the adsorbate atoms optionally grouped by
enclosing them inside parentheses-like characters (“()”, “[]” or “{}”).
The use of more than one value is possible.
molec_ctrs2 (keyword-dependent): The (groups of) atom indices in the
adsorbate to be used as second centers (ie. the atoms used to define dihedral
angles when using the internal set of angles). When a group of atom indices
is defined by enclosing them inside parentheses-like characters (“()”, “[]”
or “{}”), the second center is defined as the average of the atom’s
coordinates.
Mandatory if set_angles is set to “internal”.
Accepted values: atom indices of the adsorbate atoms optionally grouped by
enclosing them inside parentheses-like characters (“()”, “[]” or “{}”).
The number of second centers must be the same than the number of adsorption
centers/anchoring points. Groups of indices count as 1.
molec_ctrs3 (keyword-dependent): The (groups of) atom indices in the
adsorbate to be used as third centers (ie. the atoms used to define dihedral
angles when using the internal set of angles). When a group of atom indices
is defined by enclosing them inside parentheses-like characters (“()”, “[]”
or “{}”), the second center is defined as the average of the atom’s
coordinates.
Mandatory if set_angles is set to “internal”.
Accepted values: atom indices of the adsorbate atoms optionally grouped by
enclosing them inside parentheses-like characters (“()”, “[]” or “{}”).
The number of third centers must be the same than the number of adsorption
centers/anchoring points. Groups of indices count as 1.
sample_points_per_angle (optional): Number of rotations to carry out, in
the orientational sampling, per each of the three angle in the set. The total
number of rotations generated is equal to \(n^3 - n(n-1)\) where \(n\)
is the value set for sample_points_per_angle.
Accepted values: Positive integers. Default Value: 3.
Note
All possible combinations of the three different angles are generated,
leading to \(n^3\) raw configurations. However, for both, the Euler (in its
x-convention) and the internal set of angles, redundant configurations are
generated with a dependence on the rotations per angle \(n\)) equal to
\(n(n-1)\): In the x-convention of Euler angles,
\((x, 0, 0) \equiv (0, 0 , x)\). In the internal angles, when the bond
angle is flat, all the bond-dihedral angle rotations are ineffective. These
redundant configurations generated both in the Euler and Internal angles are
algorithmically filtered out.
screen_inp_file (mandatory): The input file to be used for the chosen
code in order to carry out the geometry optimization.
Accepted values: The name/path of the file, .inp for CP2K, INCAR KPOINTS (POTCAR) for VASP, dftb_in.hsd for DFTB, and .yaml for mace.
select_magns (
mandatory): Which magnitudes/quantities should be used to
select the conformers generated at the
Isolated stage.
Accepted values: “energy” or “MOI”.
set_angles (mandatory): Which set of angles must be used to perform the
orientational sampling.
Accepted values: “Internal” or “Euler”
sites (mandatory): The (groups of) atom indices in the surface to be
used as sites of adsorption (ie. where the adsorbate should be placed on the
surface). When a group of atom indices is defined by enclosing them inside
parentheses-like characters (“()”, “[]” or “{}”), the adsorption site is
defined as the average of the atom’s coordinates. A single index represents an
atop site, a group of two a bridge site and a group of three a hollow site.
Accepted values: atom indices of the surface atoms optionally grouped, by
enclosing them inside parentheses-like characters (“()”, “[]” or “{}”).
The use of more than one value is possible.
surf_ctrs2 (keyword-dependent): The (groups of) atom indices in the
surface to be used as second centers (ie. the atoms used to define dihedral
angles when using the internal set of angles). When a group of atom indices is
defined by enclosing them inside parentheses-like characters (“()”, “[]” or
“{}”), the surface second center is defined as the average of the atom’s
coordinates.
Mandatory if set_angles is set to “internal”.
Accepted values: atom indices of the surface atoms optionally grouped, by
enclosing them inside parentheses-like characters (“()”, “[]” or “{}”).
The use of more than one value is possible.
Accepted values: The name/path of the file.
surf_normal_vect (optional): The direction vector perpendicular to the
surface. This is the direction towards where the adsorbate should be placed
above the surface. The surface normal vector can be automatically guessed
using the ASANN method. This is specially useful for stepped/kinked surfaces
or nanoparticles, where the surface normal has to be defined locally.
Accepted values: a three-dimensional vector (eg. “(1, -2, 4)”), “x”, “y”, “z”,
“-x”, “-y”, “-z” or “auto”. Default value: auto.
use_molec_file (
optional): Instead of carrying out the
Screening
stage using the molecules obtained in
Isolated. It uses a single
conformer provided with a coordinates file.
Accepted values: The name/path of the file. Default value: False.
Note
When using VASP or CP2k for screening, an xyz file is created for each configuration generated by DockOnSurf. In each xyz file, input related information (site, molec_ctr, the number of performed angular “corrections”, the isolated conformer used and the angles) are stored in the header. This gives the user traceability between input parameters and collison-free generated configurations.