Tutorials
Getting Started
Welcome to this tutorial on how to install and use DockOnSurf to help you find the most stable geometry for molecules on surfaces.
As an example, we will use DockOnSurf to investigate the adsorption of isopropanol on a (100) γ-Al₂O₃ surface using VASP.
Goals of the Tutorial
This tutorial will have three main goals:
Installing DockOnSurf: We will cover how to install DockOnSurf from the Git repository and initialize the Conda environment.
Creating Input Files: You will learn how to build simple DockOnSurf input files for the different run types implemented in the software.
Practical Application: Finally, we will use DockOnSurf to perform various types of geometric optimization series, including isopropanol conformer sampling, followed by the generation and optimization of the adsorbate-surface geometry for isopropanol on the γ-alumina surface.
Installing DockOnSurf
Step 1: Clone the Repository and Set Up the PATH
First, download the DockOnSurf Git repository:
git clone https://github.com/your-repository-url/dockonsurf.git
Next, add the DockOnSurf directory to your PATH. For example, you can add this line to your .bashrc file:
export PATH="$HOME/dockonsurf:$PATH"
Replace /PATH/dockonsurf with the actual path to the DockOnSurf directory on your system. Reload your shell to apply the changes:
source ~/.bashrc
Step 2: Install Conda
If you haven’t already installed Conda, you can use either Miniconda or Miniforge:
Miniconda: https://docs.anaconda.com/miniconda/install/
Miniforge: https://conda-forge.org/download/
Once installed, open your terminal. On a UNIX system, you should see (base) at the beginning of your prompt, indicating that Conda has been successfully installed:
(base) user@hostname:~$
If (base) does not appear, you may need to add Conda initialization to your .bashrc.
Step 3: Create and Activate the Conda Environment
Navigate to the examples directory in DockOnSurf:
cd /PATH/dockonsurf/examples
Create the Conda environment using the provided .yml file:
conda env create -f dockonsurf.yml
Follow the instructions in the terminal to complete the setup. After the installation, activate the new environment:
conda activate dockonsurf
Your terminal should now show (dockonsurf):
(dockonsurf) user@hostname:~$
This means you have successfully installed and activate the correct Python environment compatible with DockOnSurf. You are now ready to run the software. If you are not familiar with python environment you can take a look at : https://docs.conda.io/projects/conda/en/latest/user-guide/tasks/manage-environments.html
Creating Input Files
In this tutorial, we will present examples for the three different types of runs that are feasible with DockOnSurf using VASP.
We highly recommend reading the section Input File Reference Manual to gain a comprehensive understanding of all the keywords used in DockOnSurf.
Additionally, while VASP input files are necessary to run the simulations, we will only provide a brief description of them. For a full understanding of VASP input files, we strongly recommend consulting the official VASP manual: https://www.vasp.at/wiki/index.php/The_VASP_Manual.
Frist, from the Git repository, navigate to the tutorial directory in DockOnSurf:
cd /PATH/dockonsurf/tutorial
Inside the tutorial directory, you will find the following subdirectories:
prep_isolated
prep_screening
prep_refinement
tools
These subdirectories contain all the files necessary to run the three different run types in sequence: Isolated, Screening and Refinement to find stable geometry of isopropanol on the (100) γ-Al₂O₃ surface. It is beyond the scope of this tutorial to explain the whole strategy of the thrre different run_types and recommand to read the dockonsurf release article : https://pubs.acs.org/doi/10.1021/acs.jcim.1c00256.
Tools Directory: The tools directory contains simple bash and Python scripts that will be used during the analysis.
prep_isolated
The isolated step explores the conformational space of the isopropanol. Inside the prep_isolated directory, you will find the following five files:
INCAR
KPOINTS
POSCAR
POTCAR
dockonsurf_isolated_vasp.inp
The last file, dockonsurf_isolated_vasp.inp, is the input file for the dockonsurf tool, which is used to perform the isolated procedure.
This input file is divided into two sections:
[Global]
project_name = Isolated_isopropanol
run_type = isolated
code = VASP
batch_q_sys = slurm
subm_script = VASP.sh
potcar_dir = /vasp/PSEUDOS_DATABASIS/XXXX/gw/potpaw/pbe/
[Isolated]
isol_inp_file = INCAR POTCAR KPOINTS
molec_file = POSCAR
num_conformers = 30
pre_opt = MMFF
The input file contains two blocks: the Global section and the Isolated section.
### Global Section
From the Global section, we see that the computation is configured to run on a cluster using the Slurm job submission system, with a script named VASP.sh. This script must be in the same directory and will handle VASP calculations.
To proceed with the tutorial, you need to ensure the input file is correctly configured for your specific job submission system and adjust the potcar_dir value.
If the POTCAR file is already provided, you can omit the potcar_dir setting.
Otherwise, ensure potcar_dir points to the correct repository containing pseudopotentials.
### Isolated Section
The number of raw conformers to be generated is set to 30 (num_conformers) in this tutorial to reduce computational cost and coherently with the size of the isppropanol. For production investigations with larger molecule, a higher number of conformers (e.g., up to 100) is recommended for better sampling of the conformational space.
Before running the geometry optimization with VASP, the isopropanol is pre-optimized using the MMFF force field.
The Isolated block also specifies the files needed to run the VASP geometrical optimization with :
INCAR: Defines the calculation parameters, settings, and convergence criteria. In this tutorial, it is configured for a simple geometry optimization to quickly obtain results while minimizing computational cost. For production runs, we recommend adjusting the level of theory for higher accuracy.
KPOINTS: Specifies the k-point sampling in reciprocal space. For a non-periodic system like a single molecule, the grid is set to 1x1x1 to avoid unnecessary sampling.
POSCAR: Contains the atomic positions and the simulation cell dimensions of isopropanol.
POTCAR: Provides the pseudopotentials for the atoms in the molecule.
prep_screening
The screening step explores the adsorbate-surface configurational space by combining the conformational sampling from the isolated step with orientational and translational sampling. Inside the prep_screening directory, you will find the following five files:
INCAR
KPOINTS
POSCAR
POTCAR
dockonsurf_euler_screening.inp
The DockOnSurf input file dockonsurf_euler_screening.inp for the screening procedure is divided into two sections:
[Global]
project_name = Screening_isopropanol
run_type = screening
code = VASP
batch_q_sys = slurm
subm_script = VASP.sh
potcar_dir = /vasp/PSEUDOS_DATABASIS/XXXX/gw/potpaw/pbe/
[Screening]
screen_inp_file = INCAR KPOINTS POTCAR
surf_file = POSCAR
select_magns = energy
confs_per_magn = 1
adsorption_height = 3
set_angles = euler
sample_points_per_angle = 3
surf_norm_vect = z
min_coll_height= 2.5
sites = 62, 59, 54, 45, (59 62), (54, 45), (45, 54, 59)
molec_ctrs = 3, 4, (3 4)
h_donor = O
h_acceptor = O
max_structures = 200
### Global Section
The Global section is similar to that in the isolated step, except that the run_type is set to screening.
### Screening Section
The Screening section introduces several new parameters. As it uses the structures generated during the isolated procedure, you do not need to specify the isopropanol POSCAR.
Note
It is possible to select a different directory from a previous computation as input for the conformers.
However, you must provide the POSCAR file for the surface, which in this case is the (100) γ-Al₂O₃ surface, as described in the study: 10.1016/j.molcata.2009.01.024.
We recommend consulting the input file reference manual for detailed descriptions of all the keywords.
Note
Python indexing starts at 0, so this must be accounted for when specifying adsorption centers/anchoring points on the surface and molecule.
To generate diverse conformers while keeping computational time reasonable, we limit the output to 200 conformers and configure the INCAR file with low-to-moderate levels of theory and accuracy for fast results. (For production calculations, higher accuracy settings are recommended.)
Finally, the KPOINTS file for VASP computations is set to a 3x3x1 grid.
prep_refinement
The refinement step processes the results obtained from the screening procedure by extracting all structures with relative energies below a specified cutoff compared to the most stable structure. These selected structures are then recomputed at a higher level of theory and accuracy.
If you navigate inside the prep_refinement directory, you will find the following files:
INCAR
KPOINTS
POTCAR
dockonsurf_euler_refinement.inp
The DockOnSurf input file contains a Global section similar to the previous steps, except for the run_type and project_name. In the Refinement section, we find:
[Refinement]
refine_inp_file = INCAR KPOINTS POTCAR
energy_cutoff = 10.0
The Refinement section specifies:
The INCAR, KPOINTS, and POTCAR input files for the VASP calculations. Since the structures are extracted from the screening procedure, no POSCAR file is required.
An energy_cutoff value (in this case, 10.0 eV), which determines the threshold for selecting strtcture to refine from the screening results.
Note
The energy_cutoff in production is usually much smaller due to higher accuracy during the screening procedure. It is set relatively high in the tutorial to ensure enough structures are available for comparison.
Thus, the main difference from the screening computations is the INCAR file, which is adjusted for higher accuracy and allows for more ionic relaxation steps to ensure well-converged results.
Practical Applications
Run Isolated
Great! Now that you are familiar with the DockOnSurf input files, let’s begin the practical part of the tutorial.
We will start by running the isolated procedure. To do so, copy all the files from the prep_isolated directory into the main tutorial directory. All the runs should be executed in the same directory, as new subdirectories will be created to store the conformers used in subsequent steps.
If you are currently in the tutorial directory, use the following command to copy the files:
cp prep_isolated/* .
This will copy all the necessary files into the tutorial directory. Once the files are in place, run the isolated procedure with the following command:
dockonsurf.py -i dockonsurf_isolated_vasp.inp
If the procedure starts correctly, you should see the following message:
Running DockOnSurf.
To check DockOnSurf activity see '/home/PATH/tutorial/dockonsurf.log'.
You can monitor the execution by checking the dockonsurf.log file, which provides updates about the process. The procedure should finish with the message:
XX-Month-XX 00:00:00-INFO: DockOnSurf finished.
During the run, a directory named isolated will be created, containing 30 subdirectories named conf_X (with X ranging from 0 to 29), corresponding to the 30 raw conformers.
Once the computations are complete, ensure that all calculations finished correctly. Navigate to the tools directory and use the extract_CPU.sh bash script. Adjust the target directory to isolated if it is not already set. This script searches for the string Total CPU time used (sec): in all VASP OUTCAR files and generates a CPU.dat file containing the name of each conformer and its associated CPU time. If a conformer’s CPU time is missing, the computation did not finish correctly.
If you want to examine the different conformers, you can also use the extract_energy.sh script, setting the target directory to isolated. This script extracts the last E0 values from each OSZICAR file and stores them in a file named E0.dat.
For example, the following image shows the lowest-energy conformer obtained during the sampling:
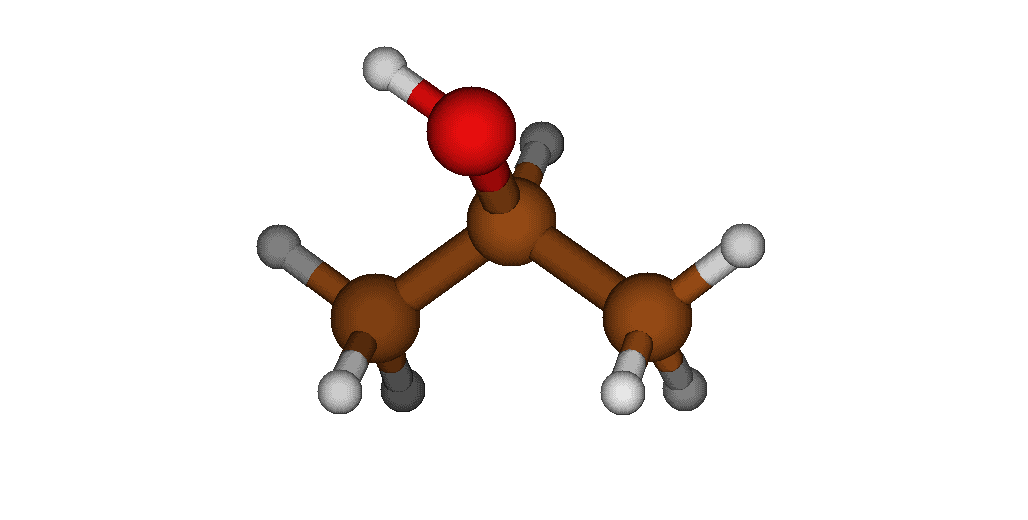
After confirming that all calculations completed successfully, you can proceed to the screening procedure.
Run screening
For the next step, you will need to replace the input files from the isolated run with the files for the screening run. If you wish to retain the isolated run files, we recommend storing them elsewhere.
To proceed with the screening run, copy the files from the prep_screening directory. Assuming you are in the tutorial directory, use the following command:
cp prep_screening/* .
This will copy the necessary files to run the screening procedure. You can launch the run with the following command:
dockonsurf.py -i dockonsurf_euler_screening.inp
You should see the following output:
Running DockOnSurf.
To check DockOnSurf activity see '/home/PATH/tutorial/dockonsurf.log'.
As before, you can monitor the run by checking the dockonsurf.log file.
The run will create 200 adsorbate-surface conformers in a new directory named screening, with subdirectories named conf_X (where X ranges from 0 to 199).
Once all the VASP calculations are finished, you can perform a quick verification using the extract_CPU.sh script to ensure all calculations completed correctly.
In this example, we observed that some conformers (around 10) did not finish correctly.
Here is an example of two conformers that did not finish correctly:
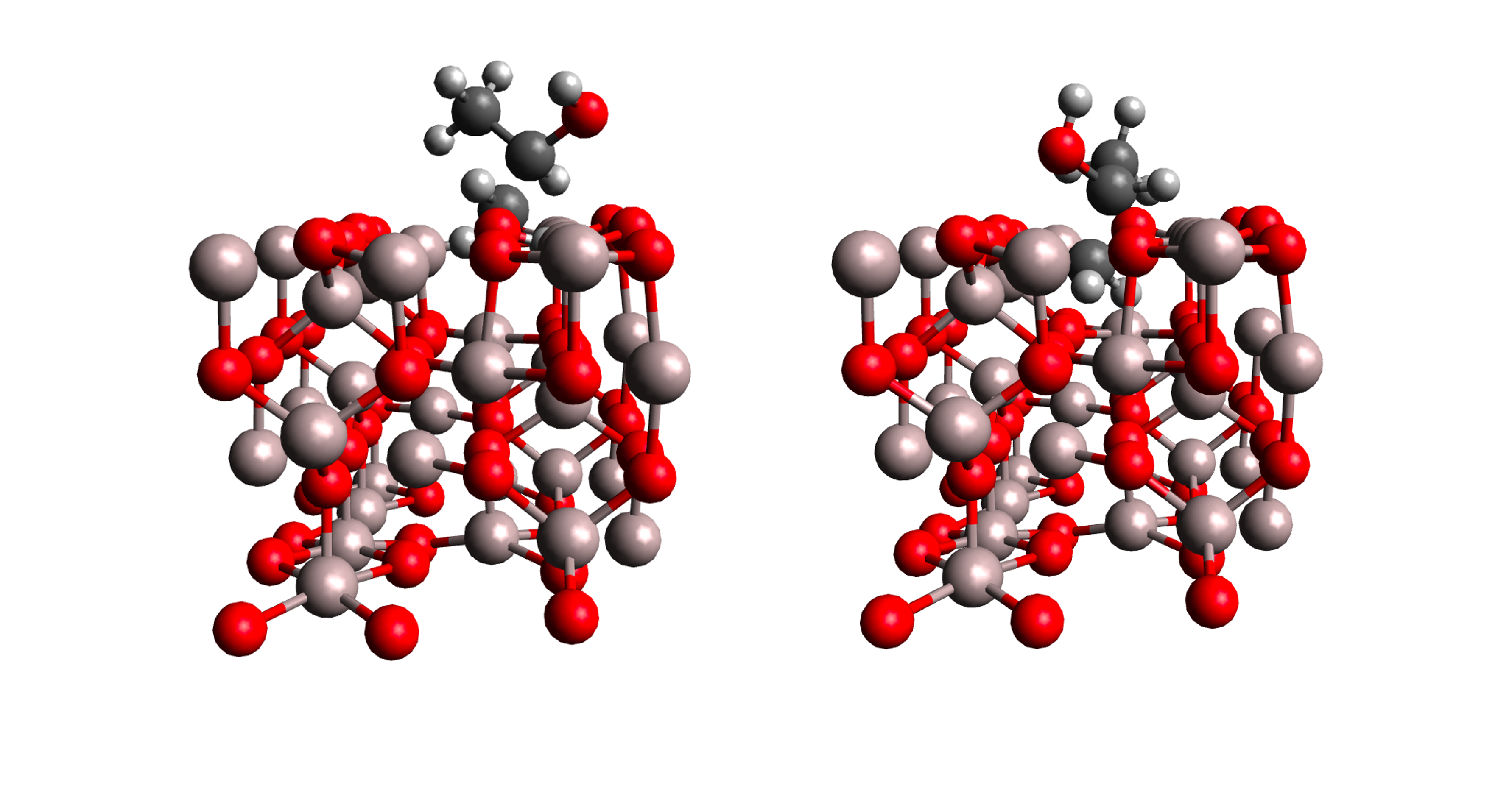
As we can see, even though the chosen molecule center (the alcohol group of isopropanol) is at the specified distance from the surface, the exploration of the conformational space with our current settings leads to overlapping structures between the adsorbate and the surface. To reduce the number of such collisions, several options can be considered. For instance, enabling the collision_threshold keyword (see documentation) could help, though it may limit the exploration of the orientational space. Morevoer, in systems where anchoring points are expected, using the internal set of angles instead of the Euler angles can improve efficiency (e.g., see the Results and Discussion section of the DockOnSurf article). After identifying the structures that did not finish correctly, they should be removed from the screening directory. Otherwise, the refinement step will produce an error message indicating that one or more runs did not complete successfully.
Next, you can analyze the energies of the adsorbate-surface structures using the extract_energy.sh script. After generating the E0.dat file, a simple Python script named extract_lowest_diff.py can be used to print the lowest-energy structure and display the minimum energy differences.
Warning
The energy from the VASP computations is reported in Hartree, while DockOnSurf uses eV as the unit of energy.
In this example, since we did not specify highly accurate or high-level geometry optimizations, large relative energy differences can be expected. In our run, we obtained the following results:
Five lowest energy differences (reference: -17964.52991662):
conf_18: Energy = -17964.52991662, Difference = 0.00000000
conf_106: Energy = -17959.06831964, Difference = 5.46159698
conf_105: Energy = -17958.45170966, Difference = 6.07820696
conf_52: Energy = -17957.52978796, Difference = 7.00012866
conf_173: Energy = -17956.20241662, Difference = 8.32750000
conf_17: Energy = -17955.92213936, Difference = 8.60777726
conf_16: Energy = -17955.85764838, Difference = 8.67226824
conf_188: Energy = -17955.54417323, Difference = 8.98574339
conf_101: Energy = -17955.23124231, Difference = 9.29867431
conf_156: Energy = -17955.06742977, Difference = 9.46248685
conf_54: Energy = -17954.88538561, Difference = 9.64453101
conf_64: Energy = -17954.28156499, Difference = 10.24835163
...
To ensure sufficient conformers for the refinement step, we set energy_cutoff = 10.0 in the DockOnSurf input file, allowing several conformers to be investigated during the refinement.
After removing, the conf_X directories that failed, and set up correctly the dockonsurf input file for the refinement, you can advance to the final step.
Run Refinement
As with the previous steps, you need to replace the input files with those from the prep_refinement directory. Assuming you are in the tutorial directory, use the following command:
cp prep_refinement/* .
This will copy the necessary files to run the refinement procedure. You can launch the refinement run with the following command:
dockonsurf.py -i dockonsurf_euler_refinement.inp
This should start the refinement run, and you will see the “Running DockOnSurf” message, along with the creation of log files.
The run will generate a folder named refinement, containing all the conf_X directories (where X corresponds to the systems meeting the relative energy cutoff). Since the level of theory and accuracy is increased in the refinement step, these calculations may take significantly longer. With the same submission script setup, the runtime for refinement can be an order of magnitude longer than for screening, as per the tutorial configuration.
Finally, verify that all calculations finished correctly. In the tutorial, all computations completed successfully but reached the maximum number of ionic relaxation steps (NSW). As a result, the structures may not be fully optimized. However, to limit CPU consumption for the tutorial, we will analyze the results obtained at this stage without further refinement.
The following figure presents ten refined structures obtained after the refinement procedure, with the lowest-energy structure highlighted by a green rectangle:
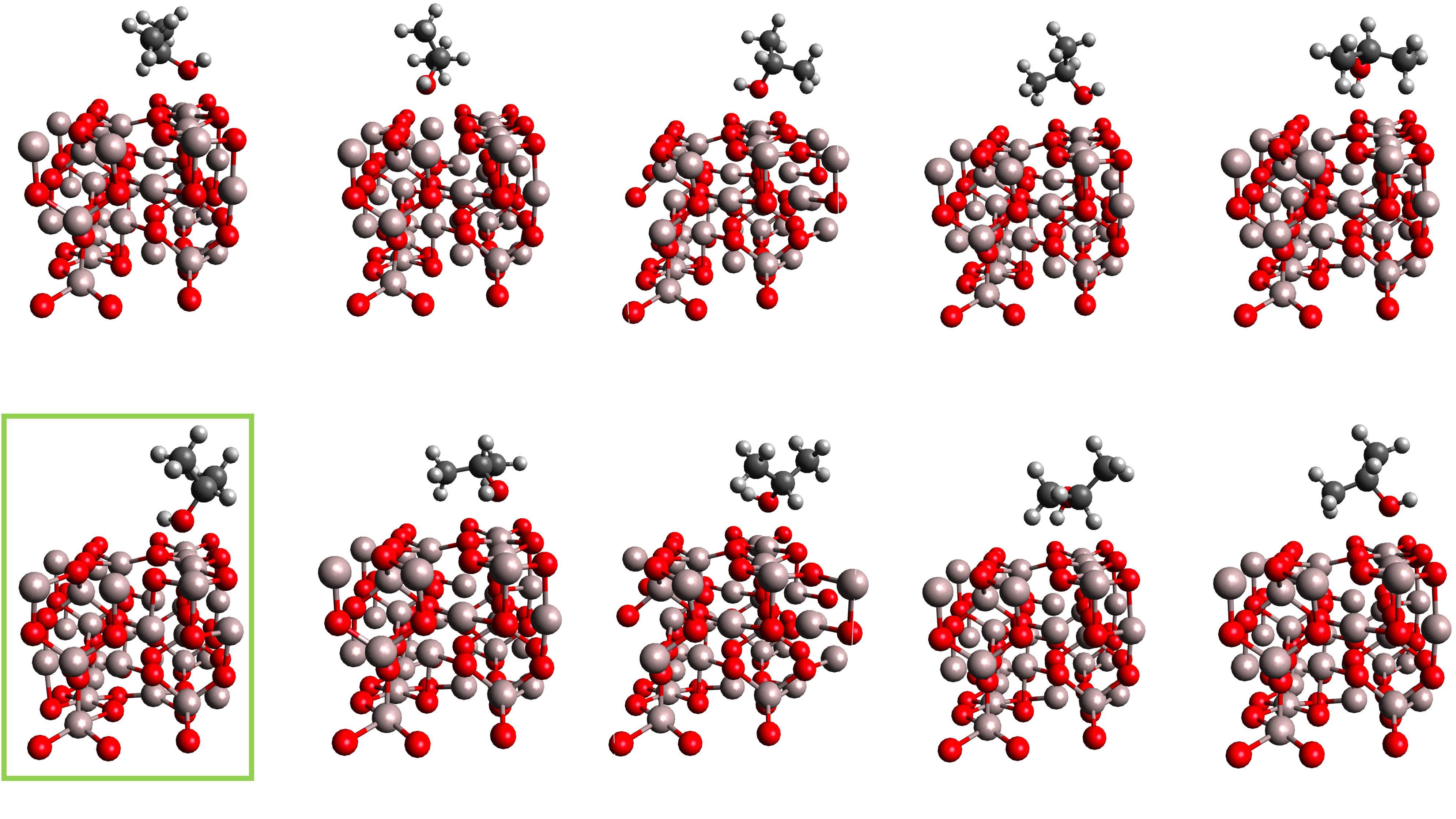
If someone is specifically interested in the adsorption of isopropanol on the (100) γ-Al₂O₃ surface, the theoretical article available at https://doi.org/10.1016/j.molcata.2009.01.024 provides a useful comparison.
From the simple series performed during this tutorial, we obtained results similar to those shown in the referenced article. This demonstrates how DockOnSurf can be used effectively to investigate stable geometries for molecules on surfaces.